
高耐熱性エポキシ樹脂の開発に向けた基礎検討
Fundamental Study on Development of High Heat Resistant Epoxy Resins
有田 和郎
Kazuo Arita
物理的耐熱性に関して,エポキシ基濃度が異なる各種アルキルフェノールノボラック型エポキシ樹脂と,平均エポキシ基数の異なるクレゾールノボラック型エポキシ樹脂を用いて架橋密度と諸物性の関係を明らかにし,それぞれを比較することで,エポキシ基濃度およびエポキシ基数がガラス転移温度や,その他の重要特性におよぼす影響をそれぞれ分離して定量的に解析した結果を紹介する。一方,化学的耐熱性に関しては,市販の様々な構造のエポキシ樹脂を用いて検証した結果を紹介する。架橋密度を高める手法では,他の機能目標が相反関係となることが明らかとなり,そのメカニズムを先人の理論を用いて解説する。更に架橋密度の増強手法では無く,新たな分子設計技術の開発の必要性を示し,特殊骨格導入による開発事例を示す。
The relationships between crosslink density and physical thermostability or other properties are shown by using cured resin of alkyl phenol novolac type epoxy resins with different density of functional group and the cresol novolac type epoxy resins with different number of average func-tional groups. The quantitative results of the influences of density and number of functional group on glass transition temperature and other important performances are introduced. On the other hand, chemical thermostability is analyzed by using commercially available epoxy resins with various structures. The elevation of crosslink density for high heat resistant causes deterioration of other performances. This mechanism is explained according to previously reported theories. Additionally, the developments of not only good physical thermostability, but also good chemical thermostability, are achieved by novel epoxy resin with special skeleton.
キーワード:エポキシ樹脂,ガラス転移温度,架橋密度,吸湿率,誘電特性
Key Words: Epoxy Resins, Glass transition temperature, Moisture absorption, Crosslink density, Dk/Df
はじめに エポキシ樹脂の歴史と特徴
エポキシ樹脂の歴史は古く,最初の発明は1938年8月にスイスのPierre Castanによって特許が許可された(スイス特許No.211116号:当時の用途は歯科材料)。本願によって,その硬化物が接着性,機械強度,電気絶縁性に著しく優れていることを見出されている。その後,1948年にCiba Geigy社が本特許を買い取りエポキシ樹脂の商業化を進めた。エポキシ樹脂が商業的にAralditeという商品名で初めて市場に紹介されたのは,1949年4月のスイスのバーゼル市で開催されたスイス貿易見本市であった。その後,Ciba Geigy社,Shell社,DOW社のクロスライセンスが成立し,エポキシ樹脂の工業的な有用性が徐々に認識された1), 2)。
エポキシ樹脂は一般的には分子内に2個以上のエポキシ基(グリシジル基あるいはオキシラン環とも呼ばれる)を持つ化合物の総称であり,数多くの種類が存在する。分子内に1個のエポキシ基を有する化合物も「反応性希釈剤」として市販されているが,三次元架橋を形成するには2個以上の反応点が必要である。
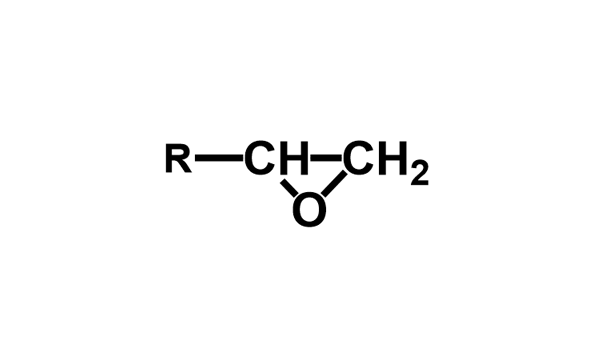
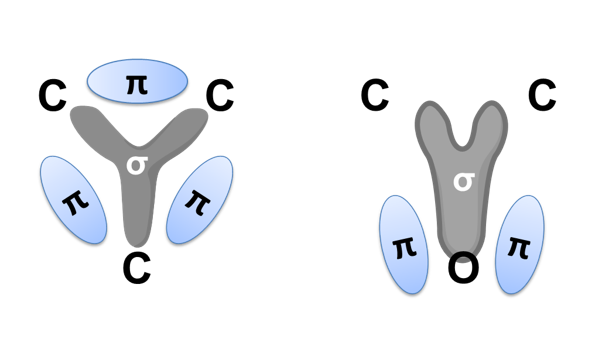
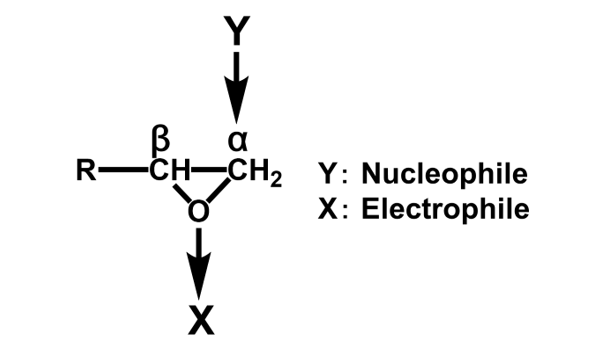
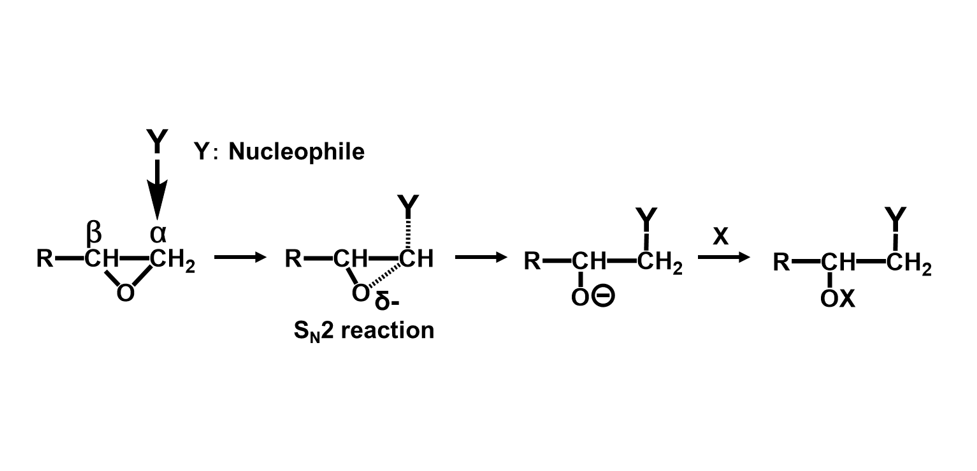
エポキシ基はFig. 1に示す通り,酸素原子1個と炭素原子2個からなる三員環構造(エポキシド)である。エポキシ樹脂の高い反応性は,この三員環構造の高いひずみエネルギーと電荷の片寄りに起因している。エポキシ基と化学構造が近似しているエチレンオキシドと,同じ三員環化合物のシクロプロパンを対比すると,後者では電荷が均一に分布しているのに対して,エチレンオキシドでは電荷が著しく酸素原子側に片寄っている(Fig. 2)3)。この電荷の片寄りでエチレンオキシドはシクロプロパンに比べて非常に大きな反応性を示す。アミノ基,フェノール性水酸基(の共役塩基),カルボキシル基(の共役塩基)などを有する求核剤は,これが炭素原子を攻撃することで反応が開始され,ルイス酸やプロトンなどの求電子剤は,これが酸素原子の攻撃を受け反応が進行する(Fig. 3)。塩基性条件下,硬化剤として一般的に用いられるアミン化合物やフェノール化合物などの求核攻撃はSN2反応であり,エポキシ基の背面攻撃で進行する。この反応は通常のSN2反応と同様にエポキシドの炭素原子の周辺の立体障害の影響を強く受ける(Fig. 3のY)。従って主に攻撃を受ける炭素原子はエポキシ基先端の2級炭素側(Fig. 3のα位)となり,α位炭素と酸素との結合が開裂しやすい(Scheme 1)4)-6)。一方,ルイス酸などを用いた場合も,開環に続く求核攻撃は(立体反転が確認されていることから)SN2反応と知られているが,遷移状態中に正の電荷を最も安定に保持できる炭素原子への攻撃が優先される。この結果として,酸性条件下では立体障害が大きい3級炭素側(Fig. 3のβ位)が求核攻撃を受け,β位炭素と酸素との結合が開裂しやすい(Scheme 2)。4)-6)
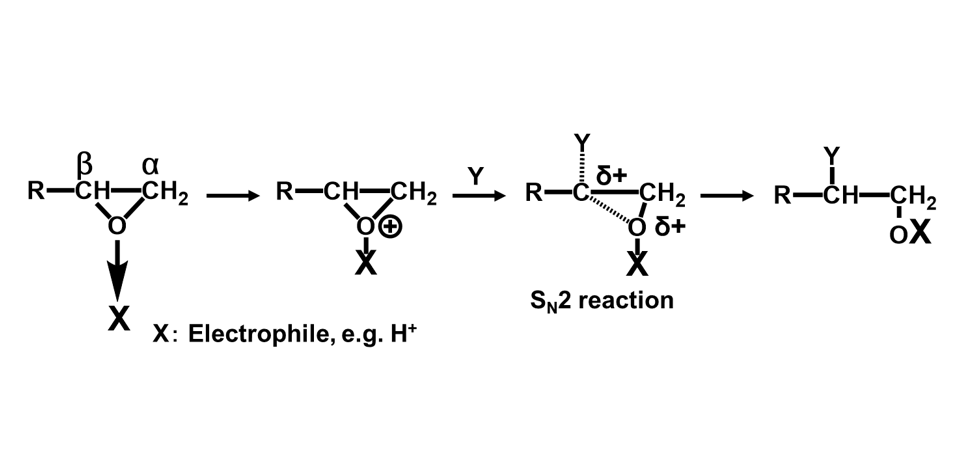
尚,余談ではあるが,IUPAC規則では一般的に置換基のある炭素側から位置番号を付けるが,エポキシ樹脂業界ではエポキシ基の先端炭素への攻撃を正常付加としており,慣例的に先端側をα炭素と呼んでいる。
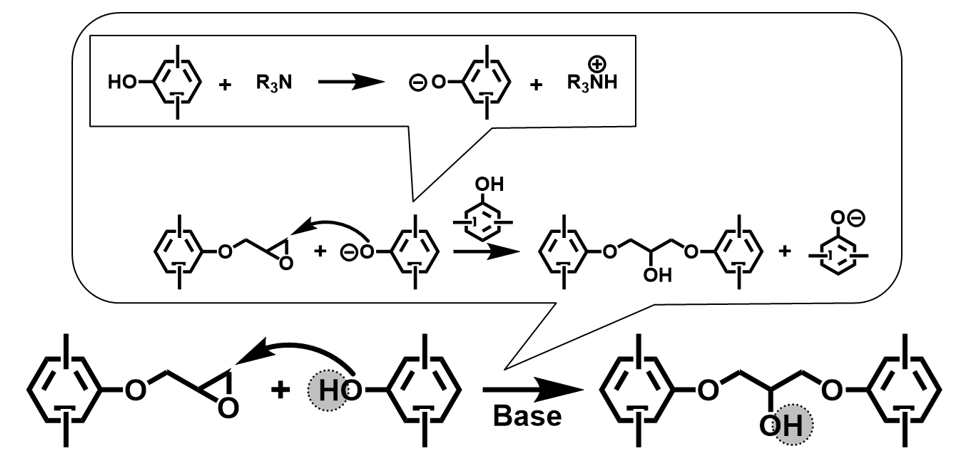
エポキシ樹脂の硬化反応の代表例として塩基性条件下でのポリフェノール型硬化剤との反応機構をScheme 3に示す。付加重合システムであり,活性水素の関与により,硬化時にエポキシ基1個から2級のアルコール性水酸基1個が生成する。この水酸基と金属やガラスとの間に発生する水素結合やファンデルワールス力によって,高い密着性が生まれると言われている。実際,同程度の耐熱性をもつ他の熱硬化性樹脂と比較すると,エポキシ樹脂の密着性は際だって優れる7), 8)。
尚,“樹脂”と名が付いているが分子量はさほど大きくなく,モノマーからオリゴマー領域にある。加熱により軟化し,更に加熱を続けると流動性を発現する。三次元架橋させ硬化物を得るためには,硬化剤となる化合物が必要であり,エポキシ樹脂そのものは,この硬化物の前駆体であることを留意して頂きたい。エポキシ樹脂と硬化剤と必要時には適切な硬化触媒を加えたうえで加熱すると,エポキシ樹脂のグリシジル基と硬化剤の活性官能基が化学反応したうえで共有結合を形成して巨大分子となり強度が発現する3)。業界によっては「(未硬化の)エポキシ樹脂と硬化剤を含む組成物」も「三次元架橋後の硬化物」もエポキシ樹脂と呼ぶ場合があるので大変紛らわしいが,特許公報の発明の名称などでは“エポキシ樹脂”,“エポキシ樹脂組成物”,“エポキシ樹脂硬化物”と区別されている例が多い。
1 緒言
ネットワークポリマーの耐熱性の指標は,2つの分類に大別できる。ひとつはガラス転移温度に代表される,機械強度や熱膨張率などの物理特性の保持についての物理的耐熱性,もうひとつは長期的に何℃まで熱分解などの化学劣化を生じることなく物性を保持できるかについての化学的耐熱性である9)。一般のパソコンなどに搭載される電子デバイスでは前者が重視されていたが,長期信頼性が重視される車載向けパワーデバイスなどの用途には,物理的および化学的耐熱性の両者が高いレベルで必要となる10)。
一方,シアネートエステル樹脂やイミド系樹脂などと比較すると,エポキシ樹脂は硬化物のガラス転移温度では劣っている場合も多いが,優れた硬化性・密着性と高耐熱性を両立できることや,ガラス転移温度以上の温度領域(ゴム状領域)で優れた物性保持力を有していることなどを鑑みると,高耐熱性エポキシ樹脂の工業的価値は非常に高いといえる。新規な高耐熱性エポキシ樹脂の開発要求は,スマートフォンやタブレットPCに代表される民生向け電子デバイスのみならず,従来はエポキシ樹脂が用いられなかったような産業分野からも高まっている。
エポキシ樹脂硬化物の耐熱性,特にガラス転移温度に代表される物理的耐熱性に関する分子構造因子としてはエポキシ基濃度,エポキシ基数,剛直性骨格,高対称性骨格,立体障害,強分極性基が挙げられている11)。エポキシ基濃度とエポキシ基数は,架橋密度に密接に関係しており,それらを高めることによって硬化物のガラス転移温度を高められる。剛直性骨格,高対称性骨格および立体障害構造は,高温環境下で起きやすくなるミクロブラウン運動の抑制に働き,強分極性基は,エポキシ樹脂硬化物中の水酸基と水素結合を形成し,ミクロブラウン運動の抑制に機能する。従って,エポキシ樹脂の高耐熱性化の手段は,これらの構造的因子の増大を図ることである。高耐熱性エポキシ樹脂の代表格はノボラック型である。これは高エポキシ基濃度,多エポキシ基数,剛直骨格などの高耐熱性化条件を満足している。
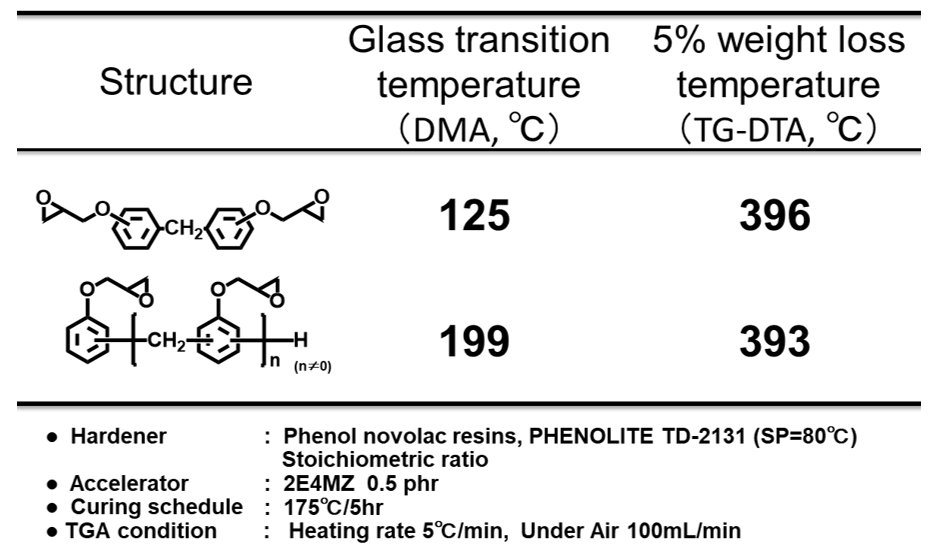
一方,熱分解温度に代表される化学的耐熱性に対するエポキシ樹脂の分子構造要因については,古くからグリシジルエーテル由来の脂肪族エーテル酸素部分や,ノボラック構造のメチレン部分が分解されやすい部位として報告されており(Scheme 4)12), 13),最近の量子計算化学においてもこの分解機構は支持されている14)。一般に熱分解温度は前記のガラス転移温度より遙かに高温であるため,ミクロブラウン運動の抑制とは別の機構で耐熱性を付与する必要がある。例えば上記したノボラック型の例のように,ガラス転移温度の向上を目的としたエポキシ基濃度の増加や繰り返し数の増加による手法では,フェニレンエーテル濃度やメチレン結合の増加に直結するため,物理的耐熱性と化学的耐熱性の傾向が一致しない例は多い(Table 1)。
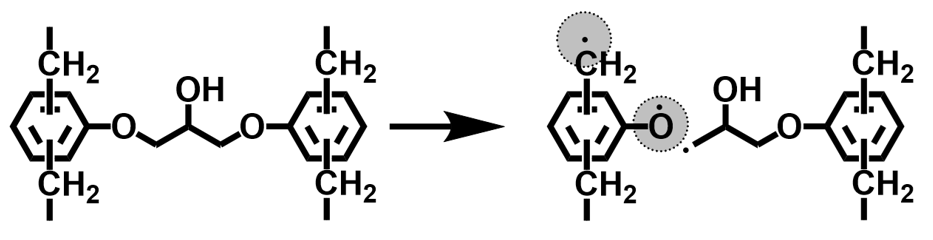
Table 1 Example of mismatch between physical and chem-ical thermostabilities
2 目的
本稿の目的は,エポキシ樹脂硬化物の耐熱性に関する基礎物性指針の構築である。得られた基礎物性指針は,目標機能を付与された新規エポキシ樹脂の分子設計の基礎となる。分子設計が明確になされれば,エポキシ樹脂の性能はさらに向上していくと考えられる。
そこで本稿では,経験的な根拠に基づき,事前に抽出した構造因子と物性との関係を定量的に把握できるような実験方法を設計して検討した。
はじめに物理的耐熱性に関して,エポキシ基濃度の異なる各種アルキルフェノールノボラック型エポキシ樹脂(Fig. 4, Table 2,平均エポキシ基数は概ね3に統一)と,1分子当たりの平均エポキシ基数の異なるクレゾールノボラック型エポキシ樹脂(Table 3, エポキシ基濃度は概ね5meq./gに統一)を用いて架橋密度とその他の重要特性の関係を明らかにし,それぞれを比較することで,エポキシ基濃度およびエポキシ基数がガラス転移温度やその他の重要特性におよぼす影響をそれぞれ分離して定量的に解析した結果を紹介する。
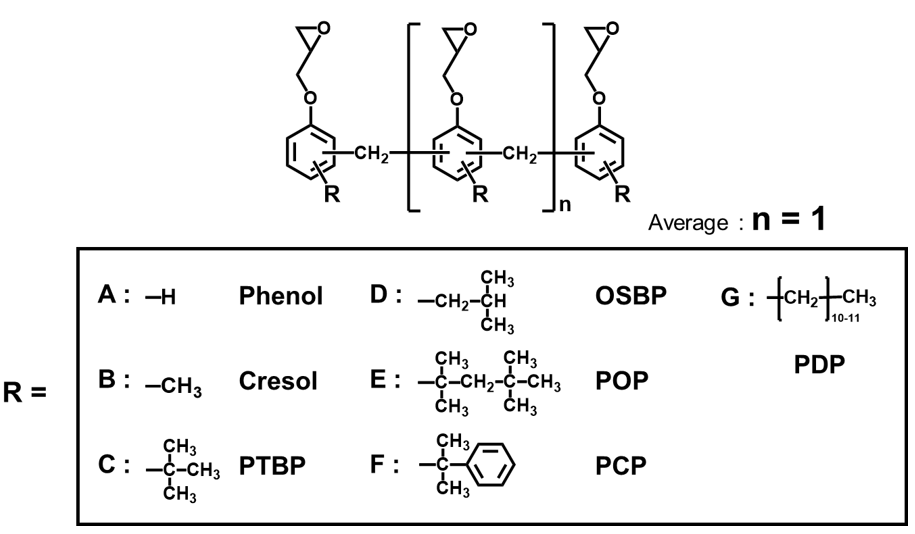
Table 2 Physical properties of phenol-novolac-type epoxy resins with various alkyl groups used for evaluation(Sample for verification of difference in epoxy group content *)
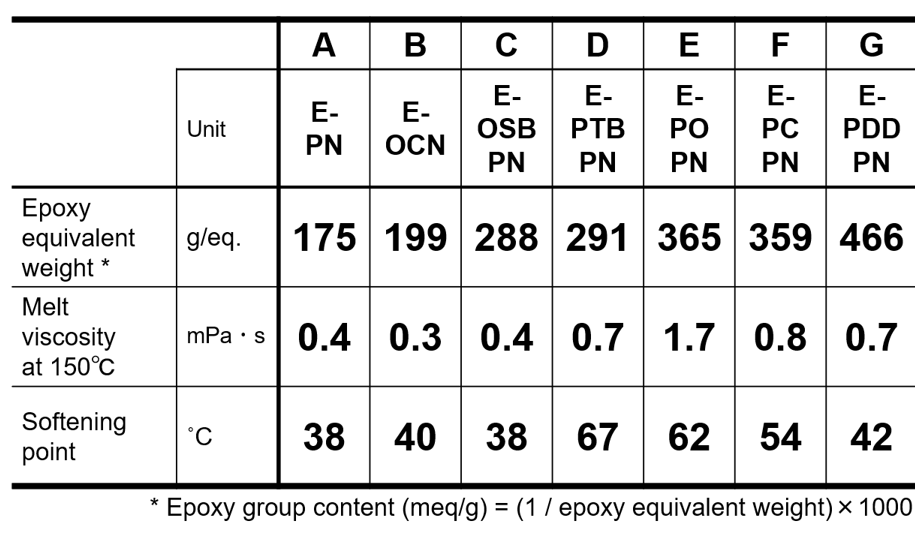
Table 3 Physical properties of cresol-novolac-type epoxy resins used for evaluation(Sample for verification of differ-ence in average number of epoxy groups)
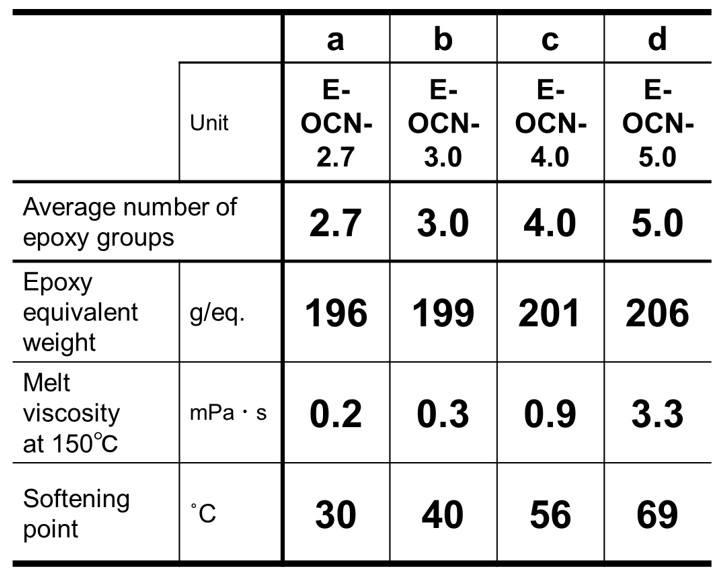
一方,化学的耐熱性に関してはTable 1に示した通り,エポキシ基数を大きく変化させても,分解温度に大きな変化が認められないことから,市販の様々な構造のエポキシ樹脂を用いて検証した結果を紹介する。
尚,前記したその他の重要特性としては,電子部材向け要求機能に直結すると考えられる,吸湿性,誘電特性および熱膨張性を選定した。
エポキシ樹脂硬化物の吸湿水分は様々な不良の原因になる。はんだ付けのためのリフロー工程では,吸湿水分が高温で急激に膨張するため(ポップコーン現象),この膨張に伴い発生した応力がクラックや界面剥離などの重大な不良を起こす。また吸湿水分はイオン性不純物の拡散を助長し,金属腐食やマイグレーションの原因にもなる。従って広範な分野における信頼性改善に対して低吸湿性エポキシ樹脂が強く求められている。
一方,信号処理速度を高めるために高周波領域(GHz帯)で作動する電子デバイスが近年,増加傾向にあるが,高周波領域の処理では伝送損失が大きくなる問題が起きている。伝送損失の低減を図る手段として絶縁材料の低誘電率化と低誘電正接化が求められる。
熱膨張性は寸法安定性に大きく影響する物性である。エポキシ樹脂硬化物は温度変化によって膨張や収縮するため,この結果として寸法変化が起こる。通常,エポキシ樹脂は熱膨張係数が小さい金属やガラスなどの基材と密着させて使われるが,厳しい冷熱サイクルを受けると寸法変化の差から接着界面に応力が起きる。その応力が機械強度や接着力を上回るとクラックや界面剥離などの致命的な不良が起こる。特に,自己発熱によりモジュール内部で急激な冷熱サイクルを繰り返すパワー半導体デバイスでは,寸法変化に各構成材料が追随できず,絶縁破壊などの重大な不良が起こる。このため,このような問題の解決手段として低熱膨張性エポキシ樹脂が強く要求されている。
このような基礎研究は,過去にも例はあるが15), 16),一般的なビスフェノールA ベースの2官能型エポキシ樹脂を用いたものが多く,多官能型エポキシ樹脂の構造因子の影響を系統的に調べた例は少ない。
3 エポキシ基濃度・エポキシ基数と諸特性の関係
3.1 ガラス転移温度
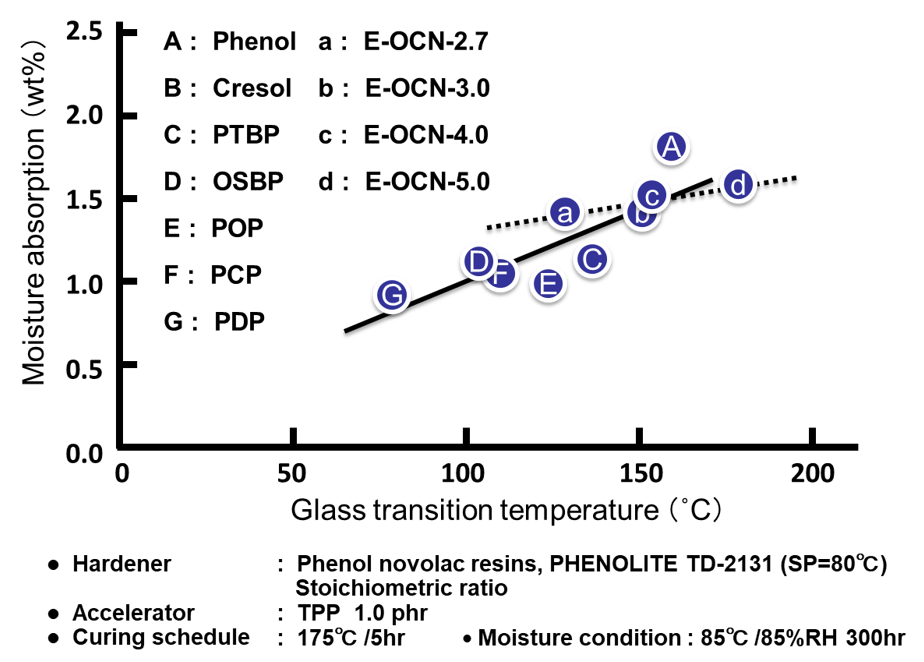
エポキシ基濃度(エポキシ当量の逆数)および平均エポキシ基数とゴム弾性理論17)から算出された硬化物架橋密度の関係をFig. 5に示す。また全てのサンプルに関してゴム弾性理論から算出された架橋密度とガラス転移温度(DMA, tanδ法, 1Hz, 昇温3℃/分)の関係をFig. 6に示す。エポキシ基濃度の増加(即ち,エポキシ当量の低下)や平均エポキシ基数の増加とともに硬化物の架橋密度が増大し,硬化物のガラス転移温度が向上することから,両者がガラス転移温度の支配因子であることが理解できる。

3.2 吸湿率
Fig. 7の左側に各種アルキルフェノールノボラック型エポキシ樹脂のエポキシ基濃度(エポキシ当量の逆数)と硬化物の飽和吸湿率(85℃/湿度85%条件下300時間後の吸湿率)の関係を示す。エポキシ基と硬化剤の反応による架橋点には2級のアルコール性水酸基が生成するため,例えば,プロットAのフェノールノボラック型エポキシ樹脂のようにエポキシ当量の低い(=エポキシ基濃度の高い)エポキシ樹脂は,架橋密度の高い硬化物を付与すると同時に,硬化物中の水酸基濃度が高くなり,吸湿率が上昇する(一方,プロットGなどは水酸基濃度が低くなり,吸湿率が低下する)。次にFig. 7の右側に平均エポキシ基数の異なるクレゾールノボラック型エポキシ樹脂の飽和吸湿率の関係を示す。Fig. 7より平均エポキシ基数と飽和吸湿率には正の相関があることが分かる。
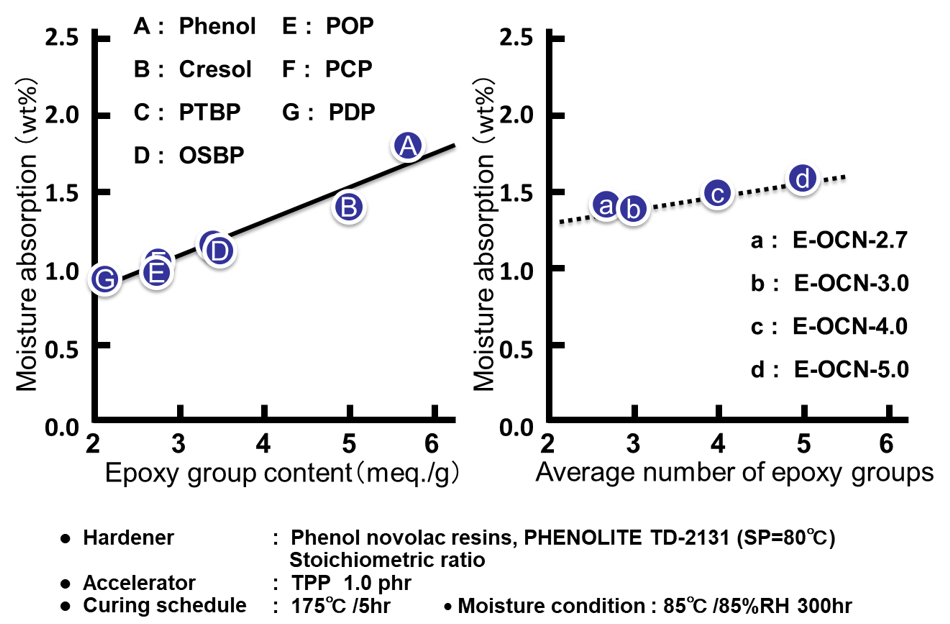
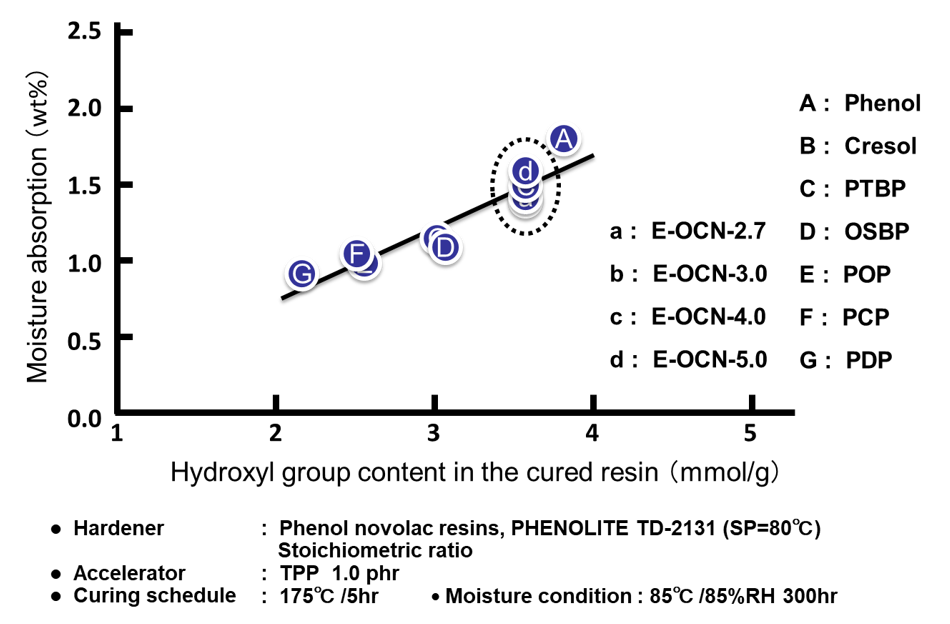
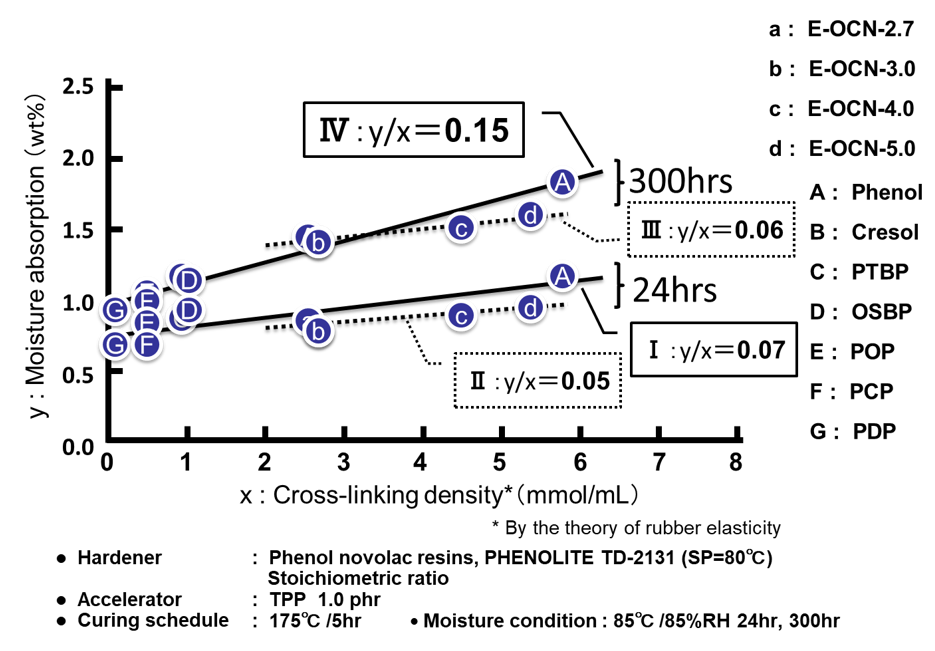
Fig. 8に全てのサンプルに関する硬化物の水酸基濃度と飽和吸湿率の関係を示す。尚,硬化物中の水酸基濃度は,硬化前のエポキシ樹脂が有するアルコール性水酸基と,硬化時に生成するアルコール性水酸基の合計とした。前者は,エポキシ樹脂を合成する際に,原料のフェノール性水酸基とエポキシ基との副反応により生成する水酸基であり,理論エポキシ当量と,実測エポキシ当量の差から算出した。後者はエポキシ樹脂と硬化剤が架橋反応する際に生成する水酸基であり,配合比率から計算した。エポキシ基濃度を変化させたプロット(A~Gの7点)は先に説明した通り,硬化物中の水酸基濃度の増加に伴い吸湿率が上昇することが分かる。一方,平均エポキシ基数を変化させたプロット(a~dの4点)は,繰り返し単位毎に官能基を有するためエポキシ基濃度はあまり変わらず,硬化物中の水酸基濃度も同じである(Fig. 8の点線で囲った部分)。Fig. 8を観る限り,a~dの吸湿率の変化は測定誤差の範囲とも見て取れるが,以下の検証から硬化物中の自由体積の変化に伴う有意差と判断している。Fig. 9にエポキシ基濃度および平均エポキシ基数を変化させた場合の硬化物の架橋密度と吸湿率の関係を示す。両図ともに架橋密度と吸湿率に非常に良い相関が認められる。ここで各プロットの傾き(架橋密度に対する吸湿率の変化率)に注目した。架橋密度に対する初期(24時間)吸湿率(Fig. 9の下段の2つのプロット)の傾きは,エポキシ基濃度を変化させた場合(傾きⅠ)も平均エポキシ基数を変化させた場合(傾きⅡ)も,大きな差は認められず,硬化物中の水酸基濃度の変化の有無の影響はほとんどみられなかった。これに対し,架橋密度に対する飽和(300時間)吸湿率(Fig. 9の上段の2つのプロット)の傾きは両者で異なった。エポキシ基濃度を変化させた場合(傾きⅣ)の傾きは,平均エポキシ基数を変化させた場合(傾きⅢ)の傾きの約2倍であった。更に,これら4つの傾きを比較すると,エポキシ基濃度を変化させた場合の飽和吸湿率の傾きⅣのみが大きな値を示し,これ以外の3つのプロットの傾き(Ⅰ,ⅡおよびⅢ)は非常に近い値となっていることが分かった。
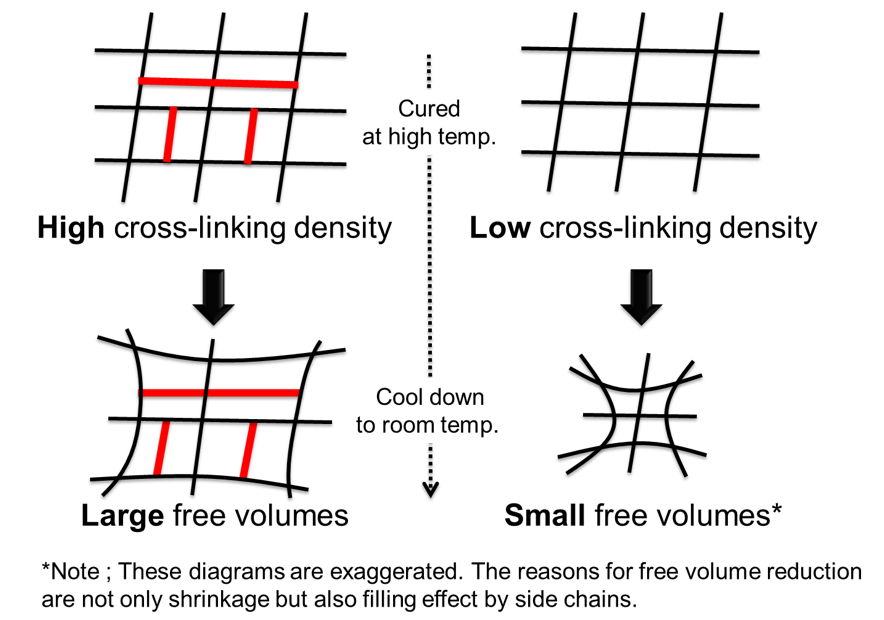
越智らの研究18)によると,エポキシ樹脂と硬化剤の配合比率を工夫することで,一般的には分離しづらい硬化物の架橋密度と水酸基濃度を独立させて検証した結果,吸湿初期の拡散係数は硬化物の架橋密度に影響を受け,飽和吸湿率は硬化物の水酸基濃度に影響を受けると報告されている。硬化物のゴム状領域の弾性率に理想ゴム弾性理論式17)を適用して求めた架橋密度と,Doolittleの方法19)によって求めた自由体積の関係から,架橋密度の高い系ほど分子間の空間が大きいと報告したうえで,この現象をゴム状領域での運動性の低い硬化物ほど冷却の際に十分な網目鎖の充填ができず,網目鎖間に多くの自由体積を残したままガラス化するためと説明している20)(Fig. 10)。即ち,架橋密度が高い硬化物ほど,分子間の空間が大きくなり,吸湿初期の拡散係数が大きな値となり,水酸基濃度が高い硬化物ほど,飽和吸湿率が上昇すると述べている。
これらと先に紹介したFig. 9の4つのプロットの傾きと照らし合わせると,①非常に近い傾きⅠ,ⅡおよびⅢを有する3つのプロットは,架橋密度の違いから生じる自由体積の変化を反映したものであり,②傾きが大きいⅣ(前記のⅠ,ⅡおよびⅢの約2倍)を有するプロットは,自由体積の変化と水酸基濃度の違いの両者を合わせた変化を反映したものと考えられる。以上より,初期の吸湿量は水酸基濃度の大小に関わらず架橋密度に強く影響を受ける一方,初期の吸湿量から飽和量までの増加分は水酸基濃度に強く影響を受けることが本研究の検証でも確認された。
Fig. 11に全てのサンプルの硬化物のガラス転移温度と吸湿率の関係を示す。架橋密度の増加は水酸基濃度や自由体積の増大を引き起こすため,高ガラス転移温度と低吸湿率は相反関係にあることが理解できる。
3.3 誘電率
エポキシ基濃度(エポキシ当量の逆数)と硬化物の誘電率について述べる。誘電率とは絶縁体の電気の溜まり易さの指標であり,溜まった電気の影響をうけて,その絶縁体に挟まれた導電部分の電気の流れが阻害を受ける。従って単位体積当たりの分極率が最も強い支配因子となり,分極率が高い構造ほど電気が蓄積されやすい。有機高分子材料の誘電率は以下に示すClausius-Mossottiの理論21)で高度な予測ができる。
Clausius-Mossotti の式
ε = [ 1+2 ( Σφ / Σν ) ] / [ 1− ( Σφ / Σν ) ]
φ:モル分極率,ν:モル体積
つまり,誘電率の決定因子はモル分極率/モル体積である。エポキシ樹脂硬化物にこの理論を当てはめた場合,水酸基濃度が誘電率への最も強い支配因子となる(Table 4)。
Table 4 Group contributions to molar polarisation and molar volume
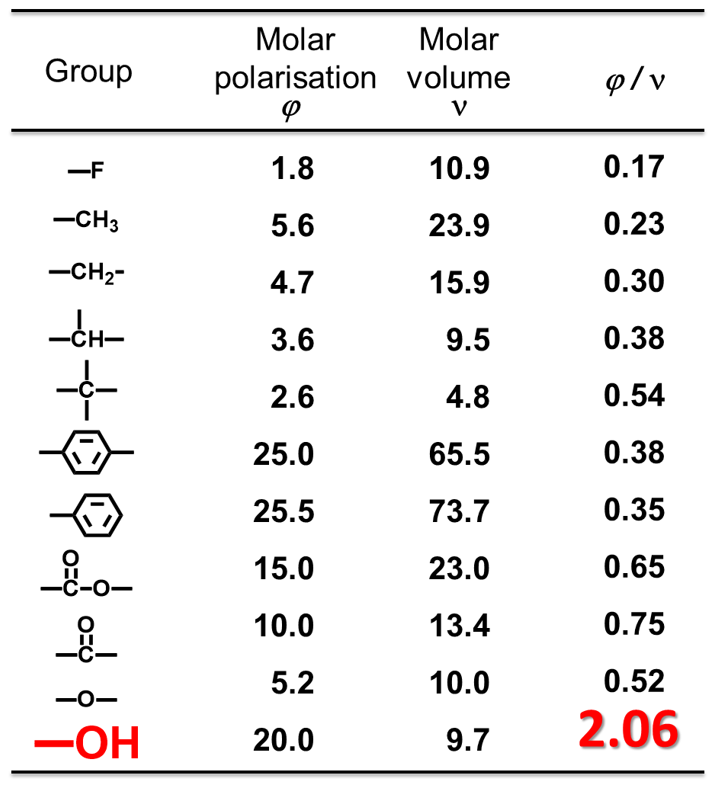
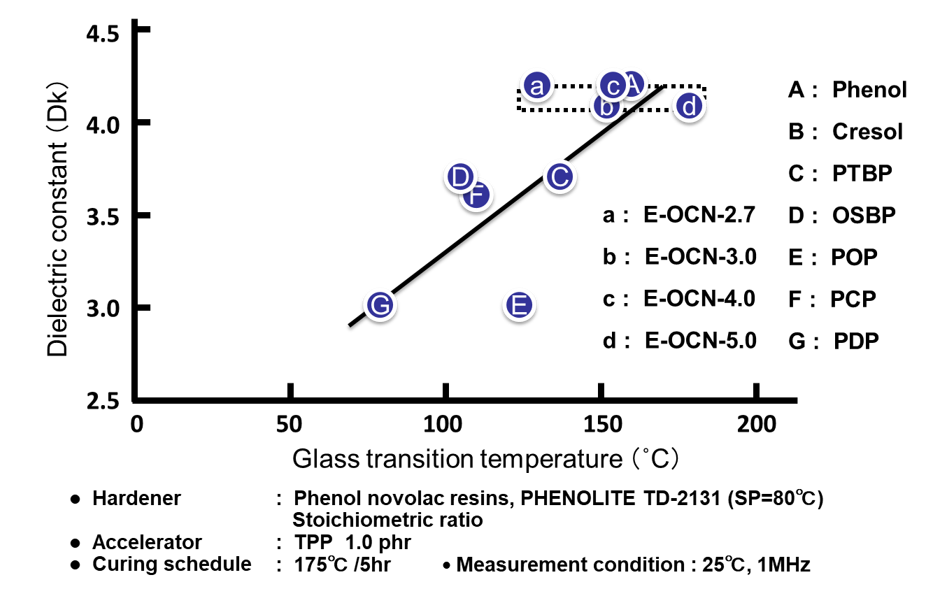
Fig. 12の左側に各種アルキルフェノールノボラック型エポキシ樹脂のエポキシ基濃度(エポキシ当量の逆数)と硬化物の誘電率の関係を示す。吸湿率の場合と同様にエポキシ当量の低い(=エポキシ基濃度の高い)エポキシ樹脂は,架橋密度の高い硬化物を付与すると同時に,硬化物中の水酸基濃度が高くなり,誘電率が上昇する。一方,Fig. 12の右側に示す通り平均核体数の違いによる硬化物の誘電率の間にほとんど差は認められない。
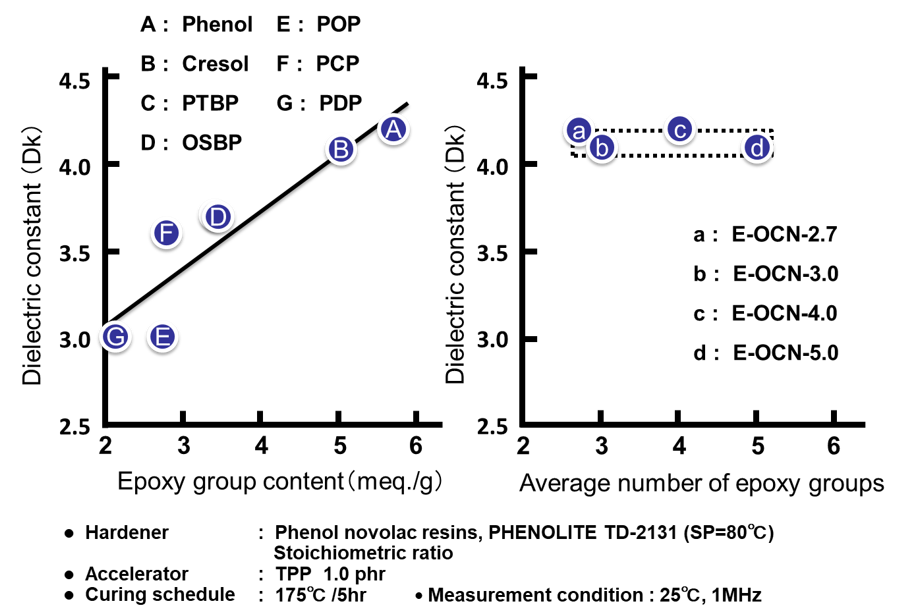
Fig. 13に全てのサンプルの硬化物中の水酸基濃度と誘電率の関係を示した。エポキシ基濃度を変化させたA~Gのプロットから,誘電率の変化は水酸基濃度の増減に因るものであることがわかる。この結果は,Clausius-Mossottiの式に,よく合致している。尚,Fのp-クミルフェノールノボラック型エポキシ樹脂は,炭素数が多く,硬化物中の水酸基濃度も低いにもかかわらず,例外的に誘電率が高い。これはクミル基が芳香族基であるために,同じ炭素数の脂肪族基と比較すると,硬化物のモル分極率が高いためと考えると説明できる。一方,平均エポキシ基数を変化させたa~dのプロットの水酸基濃度は殆ど変化しないため(Fig. 13の点線で囲った部分),電気の溜まり易さの指標である誘電率は一定の値を示す。“3.2 吸湿率”で述べた通り,吸湿率は水酸基濃度以外に自由体積を決める因子である架橋密度に影響受けるが,これとは異なり自由体積は誘電率に影響をおよぼさないことを示唆するデータと言える。
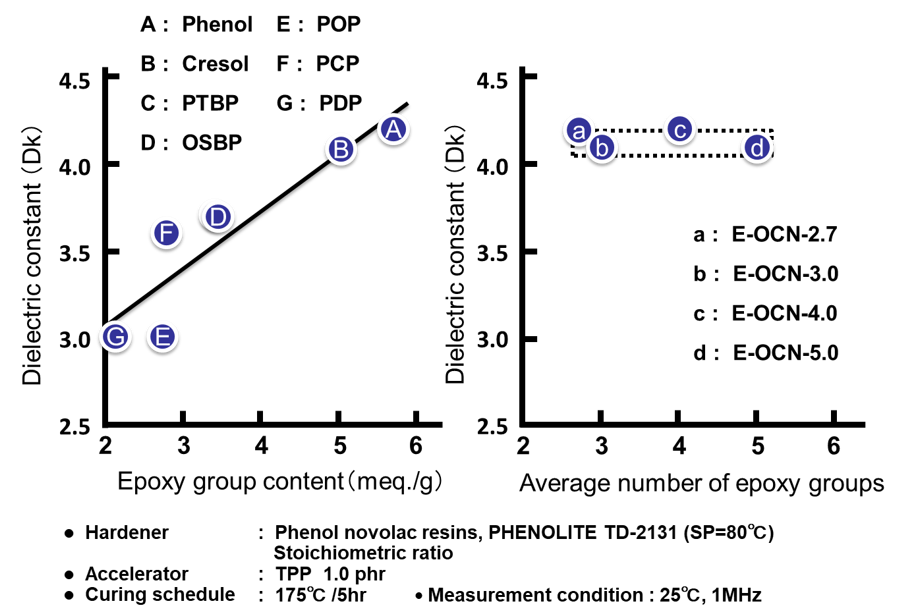
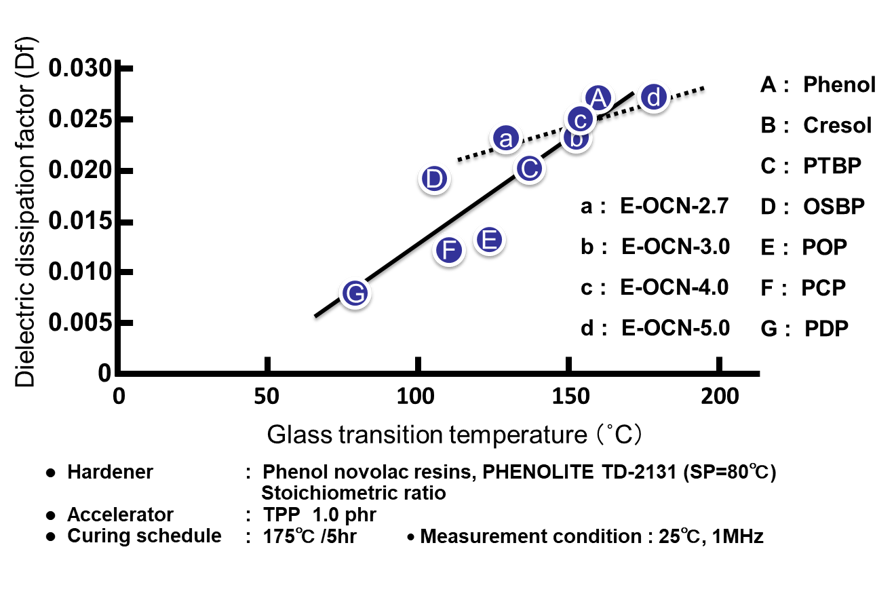
Fig. 14に全てのサンプルの硬化物のガラス転移温度と誘電率の関係を示す。エポキシ基濃度の増加に伴う架橋密度の増加は水酸基濃度の増大を引き起こすため,高ガラス転移温度と低誘電率は相反関係にある。一方,平均エポキシ基数の増加に伴う架橋密度の増加は水酸基濃度を変化させないため,この手法でガラス転移温度を向上させた場合,誘電率は影響を受けない。
3.4 誘電正接
誘電正接とは絶縁材料に交流電圧を印加した際の分子の振動性の指標である。極性基などの電場変化に追従し易い分子で構成された絶縁体は電気エネルギーが熱エネルギーに変換されてしまうので,その分のロスが発生する。従って分子の分極性に加えて分子の剛直性やパッキング性などが支配因子となり得る。本来,誘電率と誘電正接は無関係であるが,強い誘電分極部である水酸基を架橋構造中に有するエポキシ樹脂硬化物では誘電率と誘電正接に正の相関関係が成立する場合が多い。
Fig. 15の左側にエポキシ基濃度(エポキシ当量の逆数)と硬化物の誘電正接の関係を示す。この実験例では側鎖のアルキル鎖長が異なるのみで,架橋を形成する主鎖骨格は全てノボラック構造であるため,主鎖の振動性は同等であり,分子骨格の剛直性の因子は無視できると考えられる。従って誘電率の場合と同様にエポキシ当量の低い(=エポキシ基濃度の高い)エポキシ樹脂は,架橋密度の高い硬化物を付与すると同時に,硬化物中の水酸基濃度が高くなり,誘電正接が上昇する。次にFig. 15の右側に平均エポキシ基数と硬化物の誘電正接の関係を示す。Fig. 12の右側に示した誘電率との関係と異なり,Fig. 15の右側には正の相関関係が認められる。Fig. 16に示した通り,a~dのプロットの硬化物中の水酸基濃度は一定にも関わらず(Fig. 16の点線で囲った部分),誘電正接が平均エポキシ基数と相関する理由は今のころと明らかではないが,架橋密度の違いから生じる自由体積の変化が,印加した際の分子の振動性にも影響を与えているのかもしれない。実際,Fig. 17に示す通り,架橋密度と誘電正接は全てのサンプルで良い相関が認められる。
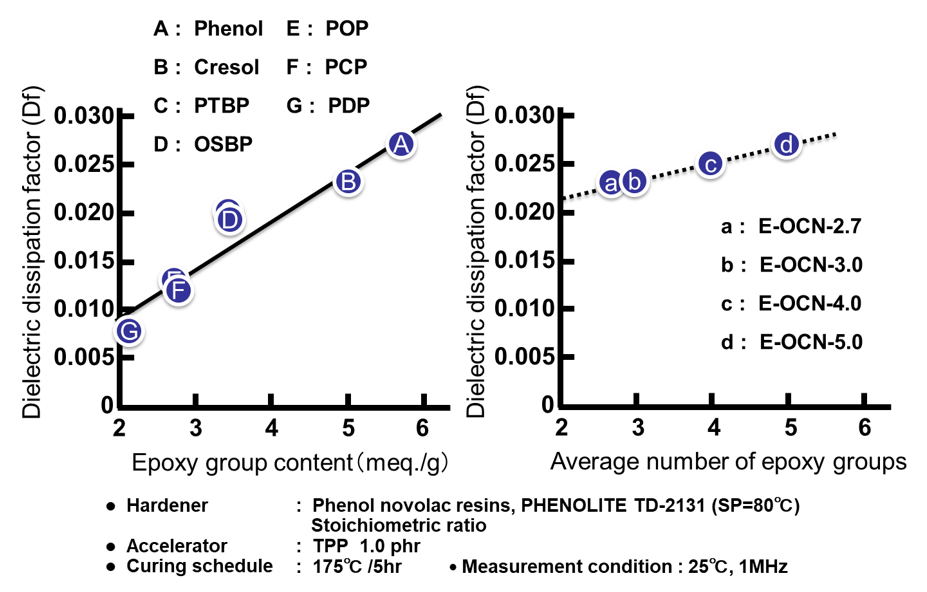
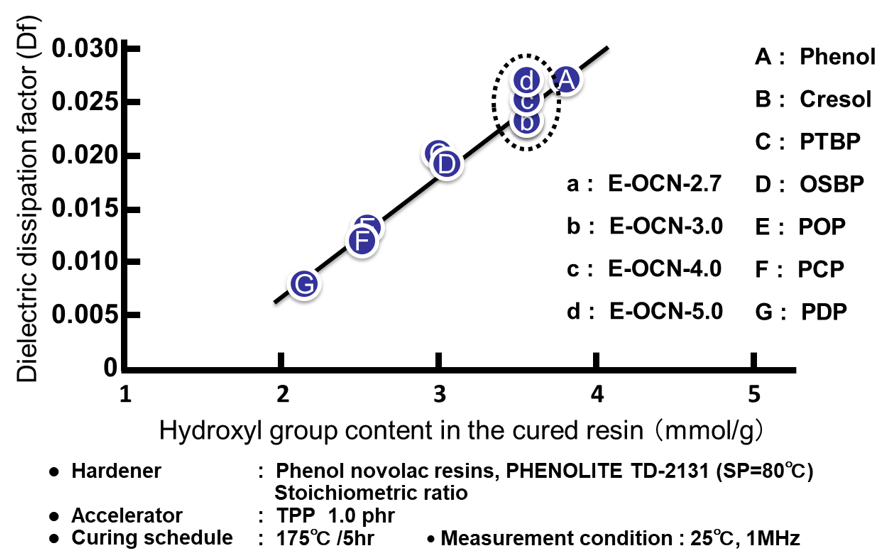
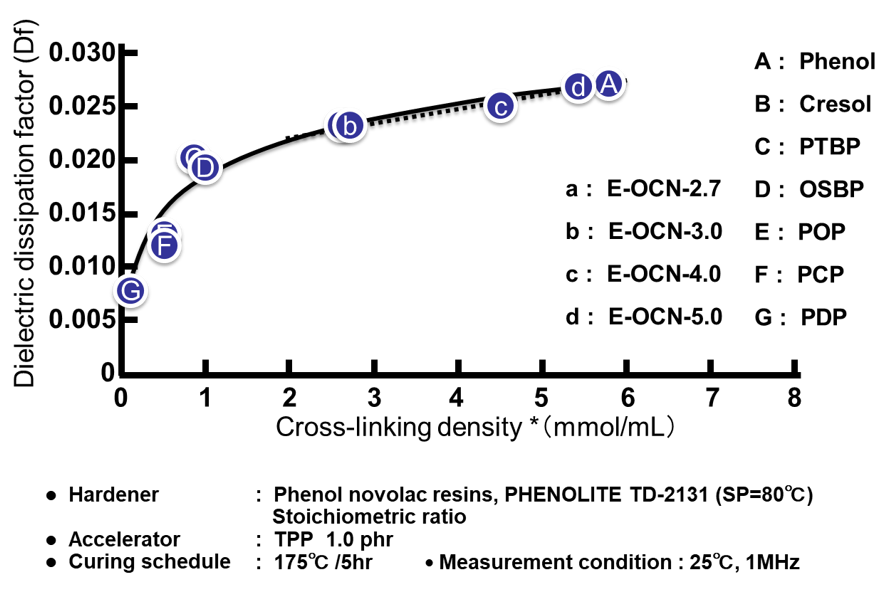
Fig. 18に硬化物のガラス転移温度と誘電率の関係を示す。架橋密度の増加は水酸基濃度を増大させるため,高ガラス転移温度と低誘電正接は相反関係にあることが理解できる。
3.5 熱膨張率
エポキシ樹脂の分子構造と熱膨張性(特にガラス転移温度以下の領域のα1)の因果関係に関しては詳しくは解明されていない。耐熱性や吸湿性などの特性に大きな変化を与えるほどに分子構造を大きく変化させてもα1にはそれほど大きな変化が現れないからである。この様な中,熱膨張率の低下には,ガラス領域においてネットワーク構造中の分子骨格のパッキング性を高め,自由体積を縮小させることで,α1を下げられるというメカニズムが提唱されている。ネットワーク構造中の分子骨格のパッキング性向上には,立体障害の小さい構造や強い分子間相互作用を誘起できる構造の導入が効果的であり,具体的にはナフタレンのような平面構造や,スルホン基などの強分極基の導入例が報告されている。22), 23), 24)
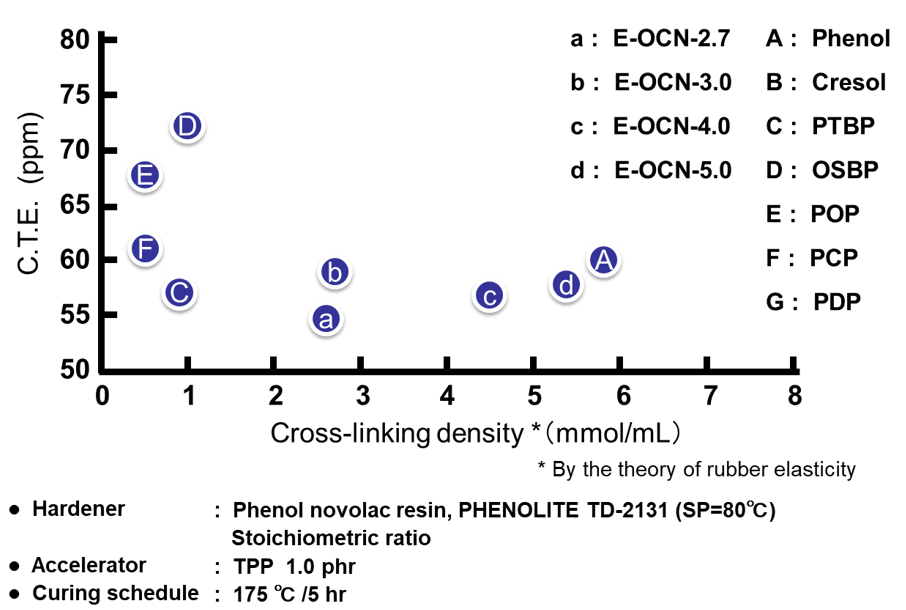
Fig. 19にゴム弾性理論から算出された硬化物の架橋密度と熱膨張率(α1)の関係を示す。架橋密度の減少は自由体積を縮小させるため,熱膨張率の低下を期待したが,我々の検討では明確な相関は認められなかった。この理由として,低熱膨張化には水素結合やvan der Waals力などの分子間相互作用やπ-πスタッキングから生じる分子配列の秩序性の付与が重要であり,今回の実験例のように分子間相互作用が無く無秩序な架橋構造をベースとした自由体積の減少では熱膨張率低下の効果を発揮しないと考察される。
4 構造と化学的耐熱性の関係
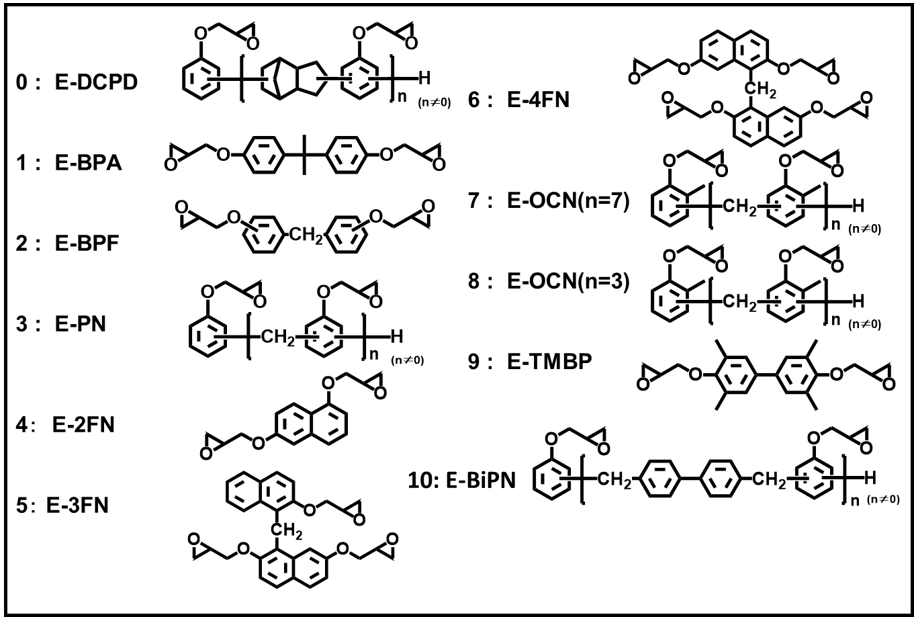
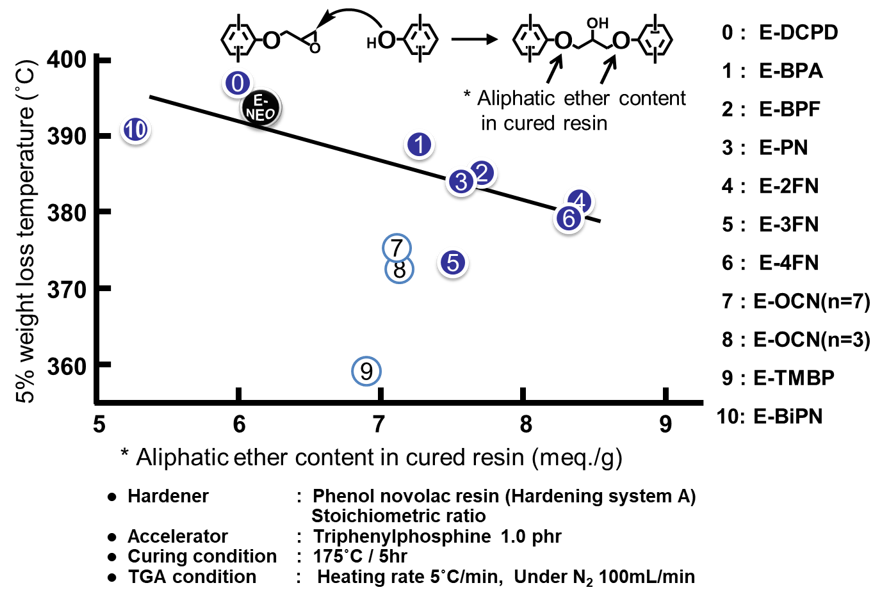
Fig. 20に示す各種エポキシ樹脂について,硬化剤として軟化点80℃のフェノールノボラックを用いて硬化物を作製し,その「5%重量減少温度」と「グリシジルエーテル基とフェノール性水酸基との架橋反応により生じる脂肪族エーテル酸素の濃度(当量配合比率からの計算値)」との関係を示した結果をFig. 21に示す。Fig. 21より,5%重量減少温度は分解の基点となる脂肪族エーテル酸素濃度におおむね依存していることが分かるが,分解し易いメチレン結合を有するナフタレン型のE-FN3やフェノール核にメチル基を有するエポキシ樹脂では,エポキシ架橋由来の脂肪族エーテル酸素濃度の寄与以上に,化学的耐熱性が低下していることが伺える。
5 高耐熱性特殊エポキシ樹脂の開発事例
著者らはこれまで2,7-ジヒドロキシナフタレンをアルカリ触媒下,200℃で反応させることにより,ナフチレンエーテルの3分子脱水反応(3量化)が選択的に進行することを見出し(Scheme 5),これを応用することで物理的耐熱性と化学的耐熱性を両立する高耐熱性特殊エポキシ樹脂の開発に成功している25)-28)。これらナフチレンエーテルオリゴマー型エポキシ樹脂(E-NEO)の硬化物は,硬化剤と架橋部位以外の構造が強固なナフタレン環と耐熱分解性に優れる芳香族性のエーテル酸素および直接結合のみであるため,高い物理的および化学的耐熱性を発現していると考えられる。
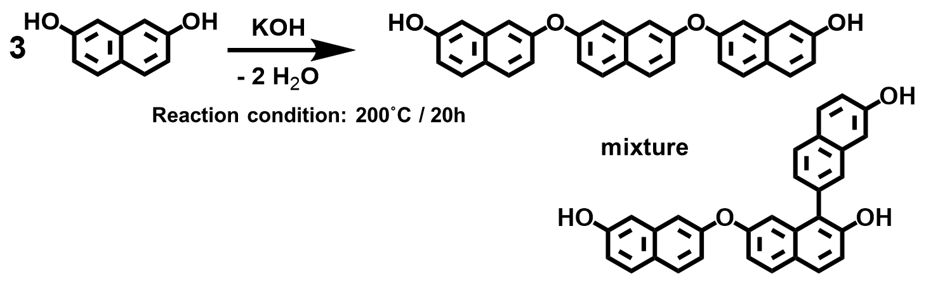
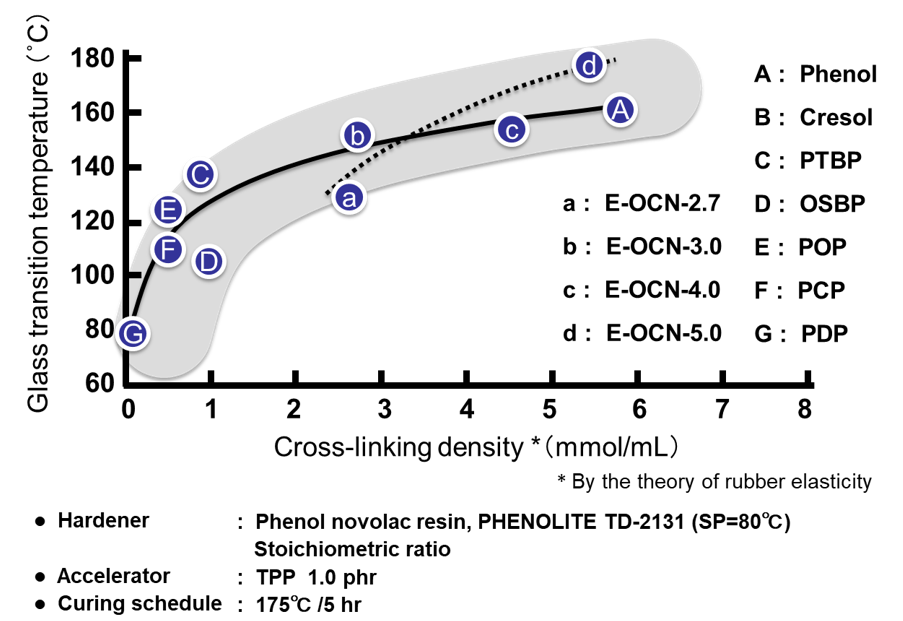
Fig. 22に本稿で用いた全ての硬化物の架橋密度とガラス転移温度の関係を示す。これらは硬化物の架橋密度とガラス転移温度の関係を,定量的に明確化したデータと言える。この定量データよりノボラック型エポキシ樹脂の場合,架橋密度の増強手法ではガラス転移温度の上限は200℃程度と推定できる。一方,E-3FNやE-4FNおよびE-NEOなどのナフタレン環骨格を有するエポキシ樹脂は高いガラス転移温度を付与している。特に,E-NEOは架橋密度とガラス転移温度の関係が特異的で,骨格の剛直性の高さが理解出来る。
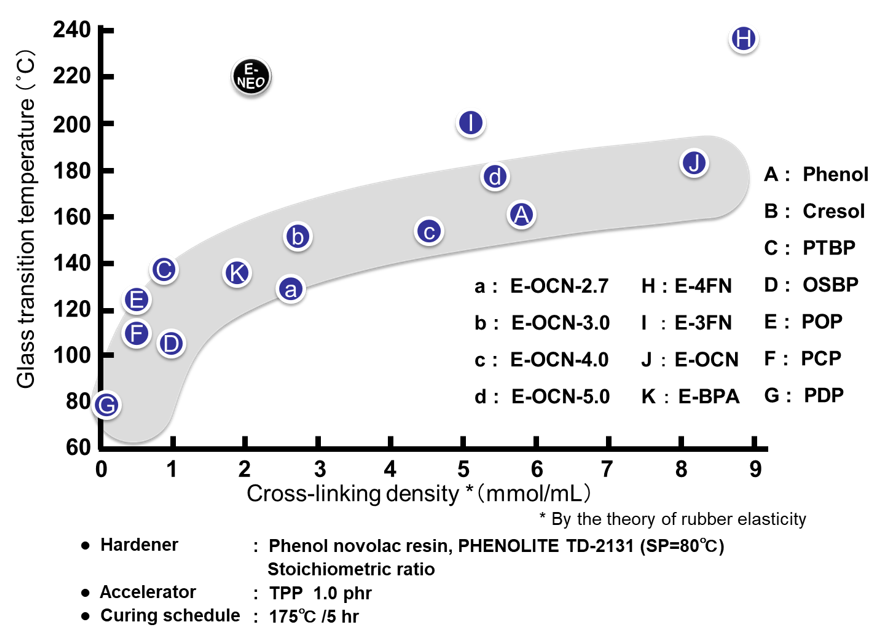
Fig. 23にガラス転移温度と相反する特性の例として吸湿率のプロットを示す。一般的に高耐熱型エポキシ樹脂は,架橋密度が高く,これまで示したように硬化物中の水酸基濃度理論や自由体積理論によって裏付けられるように吸湿特性の悪いものが多いが,図21に示した通りE-NEOの吸湿特性は通常の傾向とは反する特性を示した。E-NEOは架橋密度が低い(=架橋点に生成する水酸基濃度が低い)ながら,骨格由来の高いガラス転移温度を発現するため,高耐熱性と低吸湿性を両立する稀少なエポキシ樹脂となる。
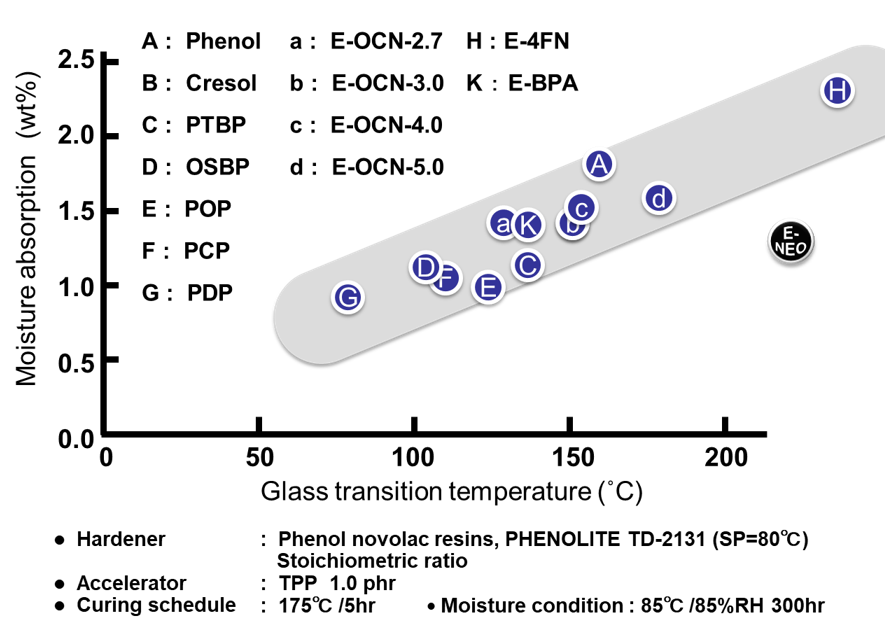
Fig. 21の「5%重量減少温度」と「グリシジルエーテル基とフェノール性水酸基との架橋反応により生じる脂肪族エーテル酸素の濃度(当量配合比率からの計算値)」との関係にE-NEOをプロットした。一般的なガラス転移温度の向上手法である架橋密度の増強を行った場合は,熱分解しやすいグリシジルエーテル由来の脂肪族エーテル酸素の濃度が高まるため,化学的耐熱性が低下する場合が多いが,E-NEOはその低い架橋密度と剛直な主鎖構造に基づいた,高い化学的耐熱性と物理的耐熱性を兼備している。
6 まとめ
エポキシ基濃度およびエポキシ基数の増加が,硬化物の架橋密度を増加させ,硬化物により高いガラス転移温度を付与することを定量的に明確化した。硬化物の諸特性に関してエポキシ基濃度の影響とエポキシ基数の影響を比較することで,硬化物の水酸基濃度と架橋密度を独立させて検証した結果,架橋密度が支配因子なのは,ガラス転移温度であり,水酸基濃度が支配因子なのは,誘電率であり,吸湿率および誘電正接は,架橋密度と水酸基濃度の両方の影響を受けることを定量的に明らかにした。これと同時に,架橋密度を高める手法では,他の機能目標が相反関係となることが明らかとなり,そのメカニズムを先人の理論を用いて関連づけた。
更に架橋密度の増強手法では無く,新たな分子設計技術の開発の必要性を示し,特殊骨格導入による開発事例を示した。
本稿は,宮澤賢史の指導のもと著者が採取したデータを用いて,小椋一郎によってまとめられた物性理論を基に,データを追加して整理したものである。ここに示した諸データが新規エポキシ樹脂の分子設計の基礎として活用されることを期待する。
本稿は,「ネットワークポリマー,36, pp.255-264 (2015).」に掲載された同名の報文を加筆修正したものである。
参照文献
- 新保正樹 編, “エポキシ樹脂ハンドブック”, p.425, 日刊工業新聞社 (1987).
- 白井博, 色材, 47, 25-35, (1974).
- 垣内弘, “新エポキシ樹脂”, p108, 昭晃堂 (1985).
- E.L. Eliel, “Steric Effects in Organic Chemistry” p.106, edited by M. S. Newman (John Wiley and Sons, New York), p.106 (1956).
- R. E. Parker, N. S. Isaacs, Chem. Rev., 59, 737-799 (1959).
- 中西香爾,黒野昌康,中平靖弘 訳,“モリソンボイド有機化学(中) 第5版”, p.912, 東京化学同人 (1989).
- 小椋一郎, “特殊半導体パッケージ(BGA) 用絶縁部材のための新規高性能エポキシ樹脂の開発研究”, 東京工業大学博士論文 (2010).
- 室井宗一, 石村秀一, “入門エポキシ樹脂”, 高分子刊行会 (1988).
- 竹市 力,“高機能デバイス用耐熱性高分子材料の最新技術,第1 章 第1 節 耐熱性プラスチックの分子設計”,pp.7-8,シーエムシー出版,2011.
- 有田和郎,エレクトロニクス実装学会誌,16, 352-358 (2013).
- 小椋一郎, DIC Technical Review, 7, 1-11 (2001).
- H. Lee:“HANDBOOK OF EPOXY RESINS, Chapter 4–12”, McGraw-Hill, Inc, 1967
- 打矢裕己,他,ネットワークポリマー,27, 151-158 (2006).
- 小松徳太郎,第63回ネットワークポリマー講演討論会要旨集, p123 (2013).
- 新保正樹, 越智光一, 小西康雄, 高分子化学, 28, 319-325 (1971).
- 新保正樹, 越智光一, 高分子論文集, 31, 124-128 (1974).
- T. Kats, A. V. Tobolsky, J. Polym. Sci., Part A, 2, 1595-1605 (1964)
- 越智光一,石井晶子,松本明彦,熱硬化性樹脂,15, 1-7 (1994).
- A. K. Doolittle, J. Appl. Polym. Sci., Appl. Polym. Symp, 34, 89-101 (1978).
- M. Ochi, K. Yamashita, M. Yoshizumi, J. Appl. Polym Sci., 38, 789-799 (1989).
- D. W. Van Krevelen:“Properties of Polymers, Chapter 11”, pp321-341, Elsevier B.V., 1997.
- 越智光一,坪内卓己,景山洋行,新保正樹,日本接着協会誌,25, 222-227 (1989).
- 小椋一郎, ネットワークポリマー, 31, 113-124 (2010).
- 大西裕一,大山俊彦,高橋昭雄,高分子論文集,68, 62-71 (2011).
- 有田和郎,小椋一郎,ネットワークポリマー,30, 192-199 (2009).
- DIC株式会社, US8729192 B2
- DIC株式会社, 特許第4285491号
- DIC株式会社, 特許第4259536号
著者紹介(執筆時)

有田和郎
DIC株式会社
R&D統括本部
アドバンストマテリアル開発センター
サイエンティスト
